

细胞衰老是对许多不同的触发因素的反应,包括DNA损伤、端粒功能障碍、癌基因激活和细胞器应激,并与肿瘤抑制、组织修复、胚胎发生和有机体衰老等过程有关。其重要性不言而喻,因此,也吸引了海内外广大研究者的注意力。那么关于衰老有什么课题的设计思路呢?我们来一起探讨一下。
接下来我们将以23年12月份发表在The EMBO Journal的一篇文章《YTHDC1通过以m6A独立的方式激活ATR来延缓细胞衰老和肺纤维化》为例,希望能给大家带来衰老相关研究的启发和灵感~

【文章题目】:YTHDC1通过以m6A独立的方式激活ATR来延缓细胞衰老和肺纤维化
【发表期刊】:The EMBO Journal
【影响因子】:IF=11.4
【发表日期】:2023.12
一、研究背景
①特发性肺纤维化(IPF)是致命疾病,但目前因细胞、分子机制不明缺乏有效治疗手段。
②DNA损伤和DNA修复活性受损,会引起肺细胞衰老和功能破坏,但DNA损伤修复机制在IPF中不明。
γH2AX信号和端粒去盖
DNA损伤修复因子的丢失
丢失或损害关键的DNA损伤反应(DDR)元件的激活,共济失调毛细血管扩张症突变和RAD-3相关(ART)
清除衰老细胞可改善肺纤维化
③YTHDC1具有良好的m6A结合基序,识别并与m6A修饰的RNA结合,以调节RNA代谢,参与多种生理和病理过程。
二、技术路线
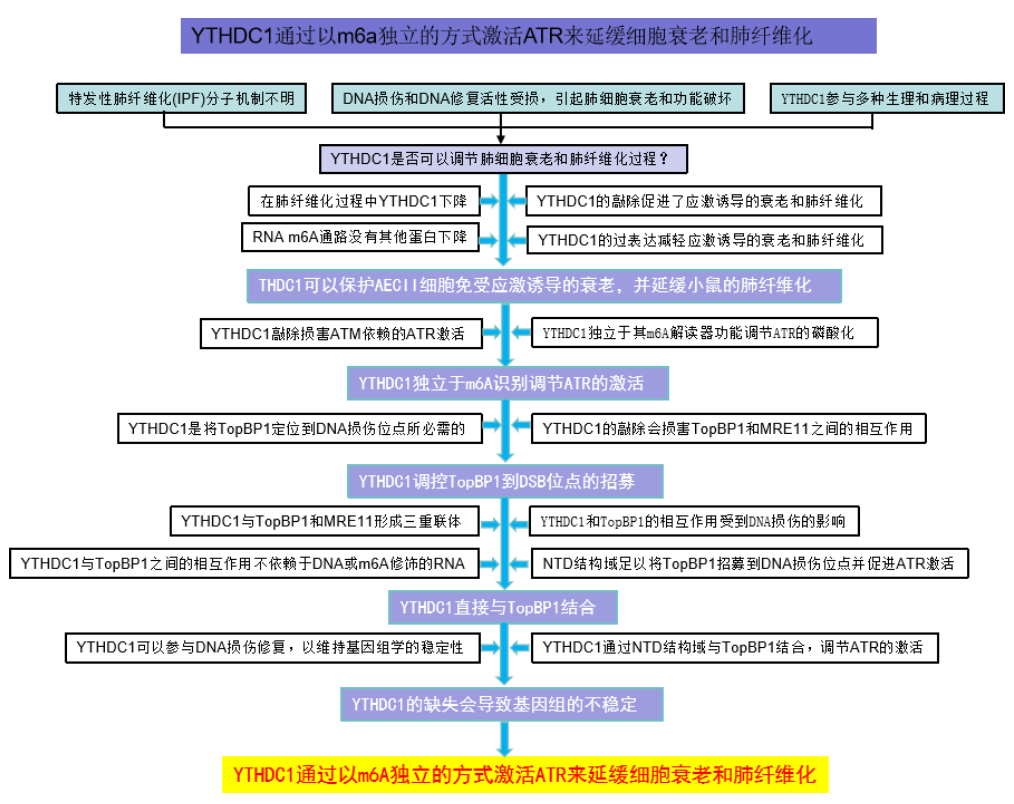
三、研究结果
1. YTHDC1的下调加重了博莱霉素诱导的肺衰老和纤维化
为了探索YTHDC1在IPF中的作用,作者通过气管内灌注博莱霉素(BLM)构建了肺纤维化小鼠模型,并采用免疫荧光法检测肺组织中YTHDC1的蛋白水平。我们从图1A~B的免疫荧光结果可以看到,YTHDC1主要在正常肺组织的肺泡上皮细胞II型(AECII,SPC阳性)细胞中表达,在肺纤维化过程中显著降低。图1C的生信数据分析显示YTHDC1在IPF病人中低表达。其他相关分析显示m6A的其它参与蛋白无变化。这说明在肺纤维化过程中,YTHDC1下降,但RNA m6A通路没有其他蛋白下降。
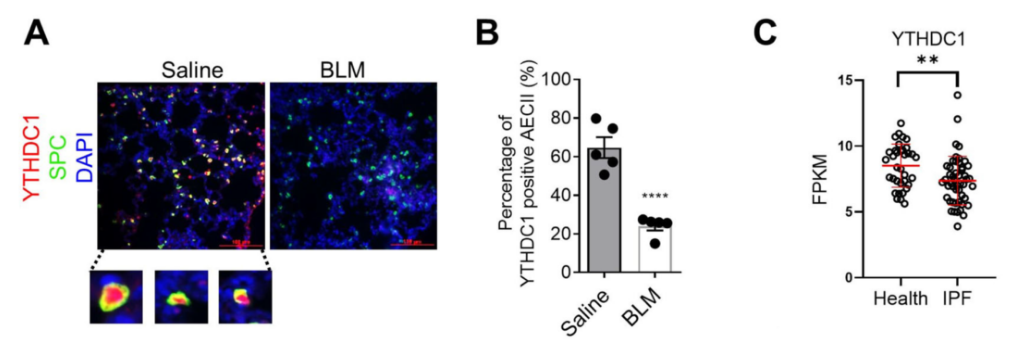
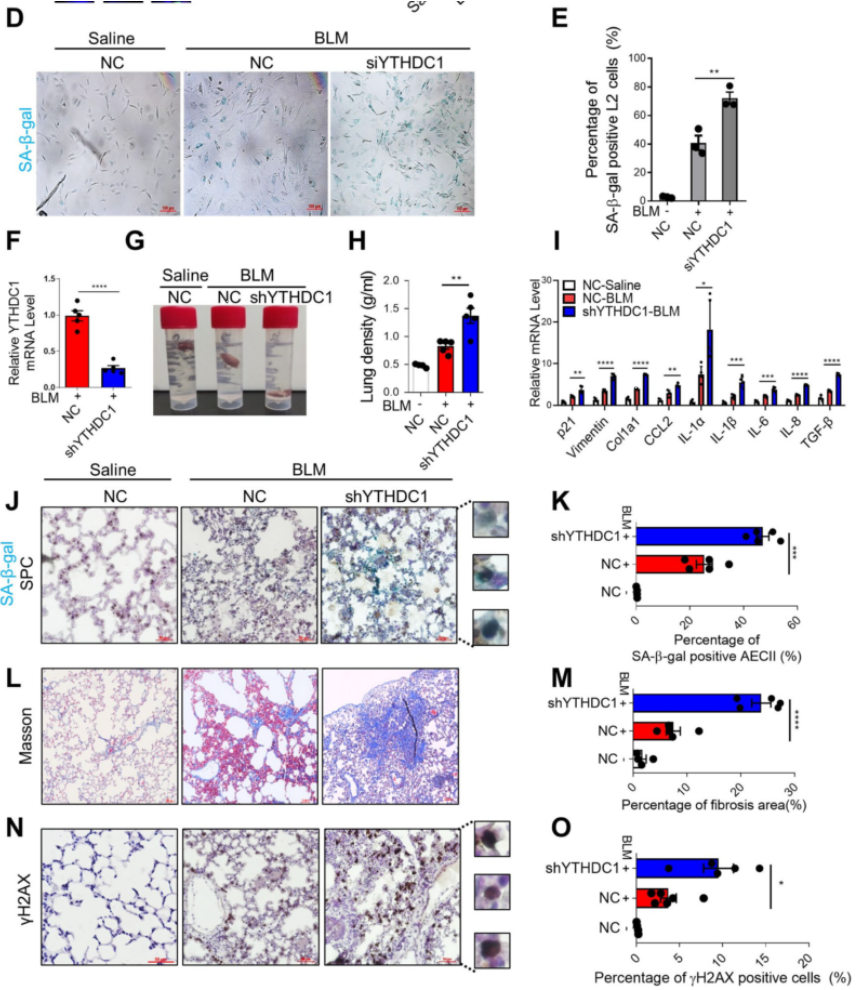
接着,作者进一步研究了YTHDC1缺失对应激诱导的细胞衰老的影响。我们从图1D~E可以看到,将YTHDC1敲除,BLM刺激后,衰老相关β-半乳糖(SA-β-gal)阳性细胞增加证明细胞衰老恶化,增殖减少。接着,作者使用腺相关病毒血清型6(AAV6)表达系统敲除小鼠肺中的YTHDC1,敲除效果如图1F所示。在7天后对小鼠进行BLM或生理盐水注射,并在图1G~O的进一步分析中发现YTHDC1的敲除显著恶化了BLM诱导的小鼠肺生理功能下降、肺衰老、纤维化和DNA损伤,这些可以从肺密度、SASP、SA-β-gal的增加、衰老标记物p21和p16 Masson的三色染色、纤维化的标志物α-SMA和DNA损伤标记物γH2AX指标得到证明。最终,这些结果表明YTHDC1的敲除促进了应激诱导的衰老和肺纤维化,这支持了YTHDC1在肺衰老和纤维化进展过程中发挥保护作用的观点。
2. 过表达YTHDC1可减轻BLM诱导的肺衰老和纤维化
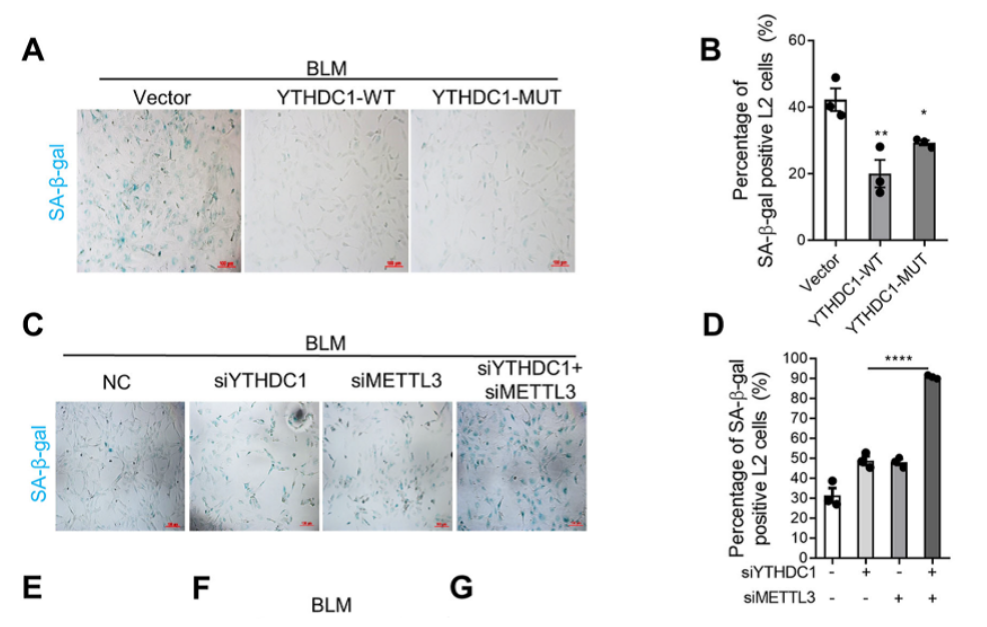
为了进一步验证YTHDC1在肺衰老和纤维化过程中的作用,作者又进行了YTHDC1过表达的实验。图2A~B显示,无论是野生型的还是突变型(没有m6A结合活性)的YTHDC1过表达,都降低了SA-β-gal、p21和p16的水平,增加ki67阳性细胞。而我们从图2C~D中可以看到,同时敲除YTHDC1和METTL3进一步加重了YTHDC1或METTL3单独缺失诱导的衰老。这表明YTHDC1可以保护细胞免受应激诱导的衰老不依赖于m6A通路。
我们从图2E可以看到,作者还使用AAV系统在肺纤维化小鼠模型中过表达野生型或突变型YTHDC1。而图2F~M的多种结果表明,过表达野生型和突变型的YTHDC1均可抑制BLM诱导的衰老和纤维化。因此我们可以得知,YTHDC1在应激诱导的肺细胞衰老和纤维化中发挥保护作用,而不依赖于其m6A识别。

3. YTHDC1独立于m6A识别调节ATR的激活
然后,作者为了验证ATM/ATR的磷酸化是否受到YTHDC1的调控,又进行了下一步实验。我们可以从图3A~C中发现,在YTHDC1敲除的细胞中,p-ATR减少,而p-ATM增多,而总ATM和ATR无显著变化。YTHDC1敲除损害ATM依赖的ATR激活。只有YTHDC1基因敲低降低了ATR的磷酸化,而其他m6A修饰相关蛋白对其没有影响,如图3D~L所示。在YTHDC1缺失的A549细胞中,YTHDC1-WT或YTHDC1-MUT的过表达均增加了ATR的磷酸化,如图3M~O所示。这些结果说明YTHDC1独立于其m6A解读器功能调节ATR的磷酸化。
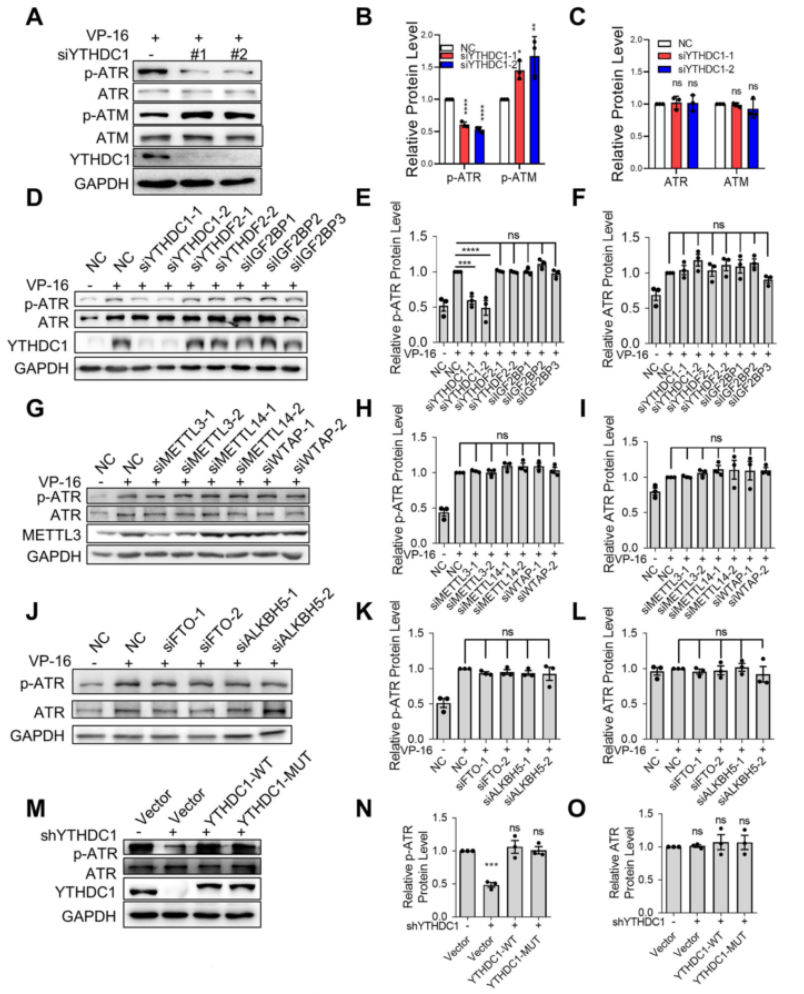
接着,为了解决“YTHDC1对应激诱导的衰老和肺纤维化的影响是否取决于ATR的激活”这个问题,作者在肺纤维化小鼠模型中检测了p-ATR,发现肺纤维化组中p-ATR病灶数和p-ATR病灶的细胞百分比均显著增加,而在shYTHDC1组中下降到正常水平,如图3P~R所示。最终结果表明YTHDC1对应激诱导的衰老和肺纤维化的影响依赖于ATR的激活。
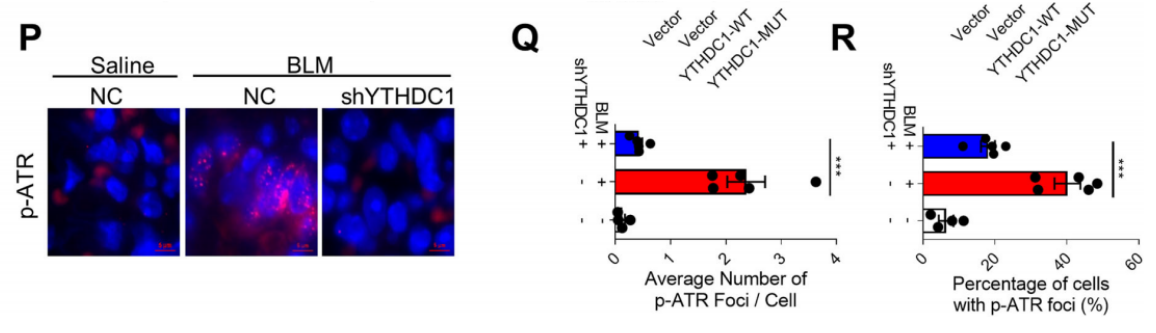
4. YTHDC1调控TopBP1到DSB位点的募集
为了探讨YTHDC1调控ATR激活的机制,作者确定了YTHDC1是否影响损伤位点上ATR激活调控因子(TopBP1、ATRIP、RAD17、RAD9A和RPA1)的募集(表示为γH2AX信号)。图4A~D显示,YTHDC1的缺失会损害TopBP1在DNA损伤位点的定位,但不影响ATRIP、RAD17、RAD9A或RPA1的募集。图4E~G显示,在VP-16处理的细胞中,YTHDC1敲除后,TopBP1核灶和共定位于γH2AX灶均减少,而TopBP1的表达水平没有变化。实验结果表明,YTHDC1是将TopBP1定位到DNA损伤位点(DSB)所必需的。
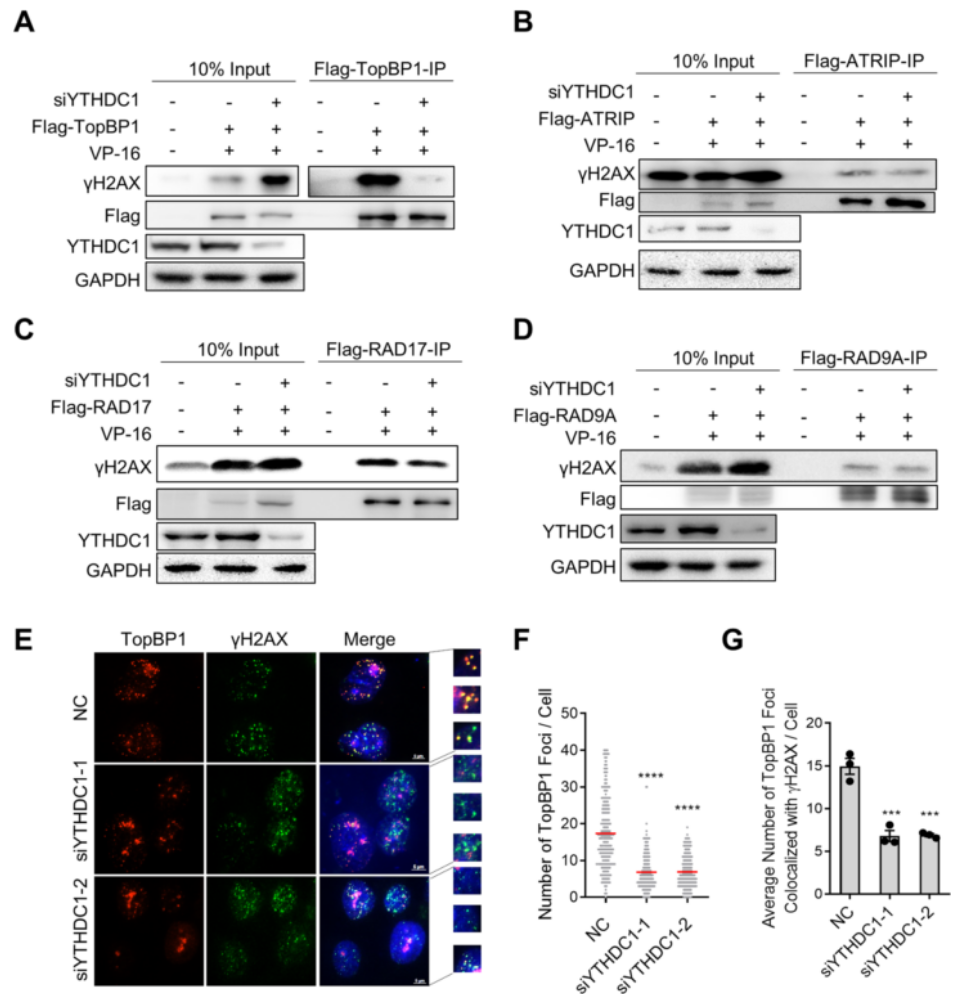
然后,作者又进一步研究了YTHDC1是否通过影响MRN复合物介导的其他方式来调控TopBP1的募集。图4H显示MRN复合物的募集不受YTHDC1缺失的影响,因为MRE11病灶不变,但TopBP1 IP沉淀的MRE11降低,这表明YTHDC1的敲除会损害TopBP1和MRE11之间的相互作用。图4I显示,同时敲除YTHDC1和MRE11对ATR活性的抑制程度等于YTHDC1或MRE11的分别敲除。从结果中可看出,YTHDC1通过促进TopBP1的募集到DSB位点来调控ATR的激活,而不依赖于m6A修饰。
5. YTHDC1直接与TopBP1结合
为了研究YTHDC1是否直接与TopBP1结合,作者在HEK293T细胞中进行了免疫共沉淀(co-IP)实验。图5A显示,在VP-16或BLM处理的细胞中,Flag-YTHDC1细胞成功拉下内源性TopBP1,内源性MRE11也被拉下。这表明YTHDC1与TopBP1和MRE11形成三重联体。图5B显示,在VP-16处理过的细胞中,Flag-TopBP1拉低了内源性的YTHDC1,表明YTHDC1和TopBP1的相互作用受到DSB的影响。图5C显示,YTHDC1-WUT可以像YTHDC1-WT一样与TopBP1结合,其相互作用不被RNase A或苯甲酰酶(BN)处理破坏,表明YTHDC1与TopBP1之间的相互作用不依赖于DNA或m6A修饰的RNA。图5D显示,在没有DNA和RNA的情况下,YTHDC1可以直接与TopBP1结合。
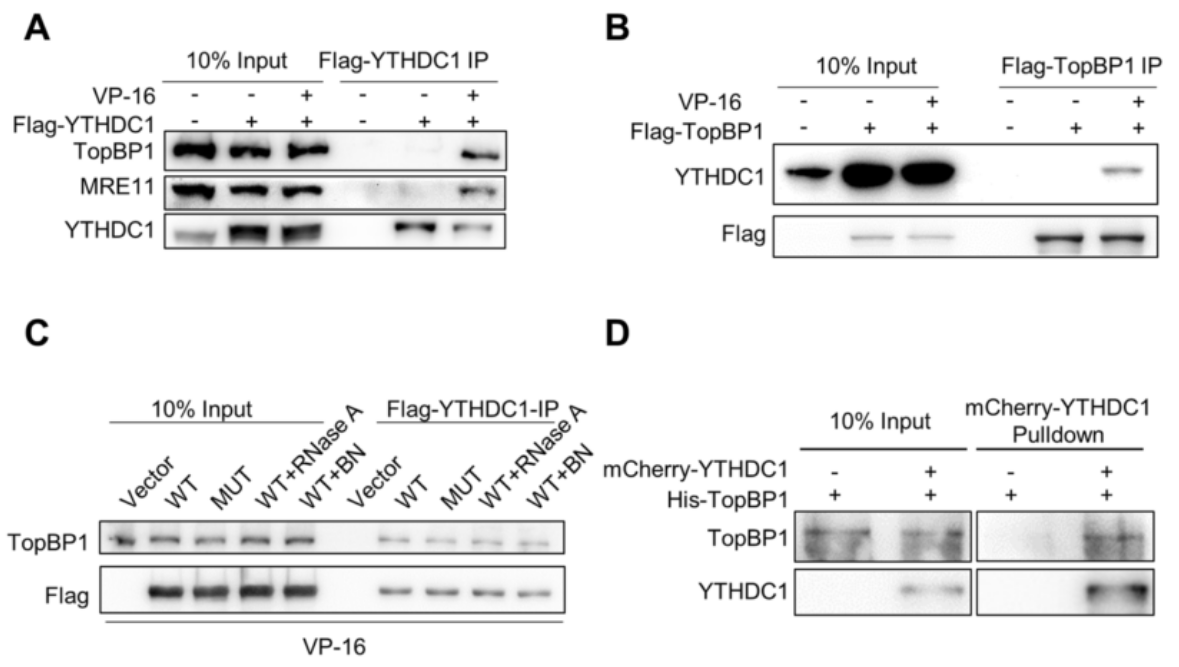
然后我们可以从图5E中发现,根据在表达flag标记的YTHDC1片段(NTD、YTH或CTD结构域)的细胞中进行的co-IP实验,TopBP1被NTD片段拉下,而不被YTHDC1的YTH或CTD片段拉下。更重要的是,MRE11也可以与NTD片段一起沉淀,这可能解释了NTD结构域足以将TopBP1招募到DNA损伤位点并促进ATR激活的结果。图5F则显示,TopBP1的BRCT结构域(1、2和3)有助于与YTHDC1的相互作用。
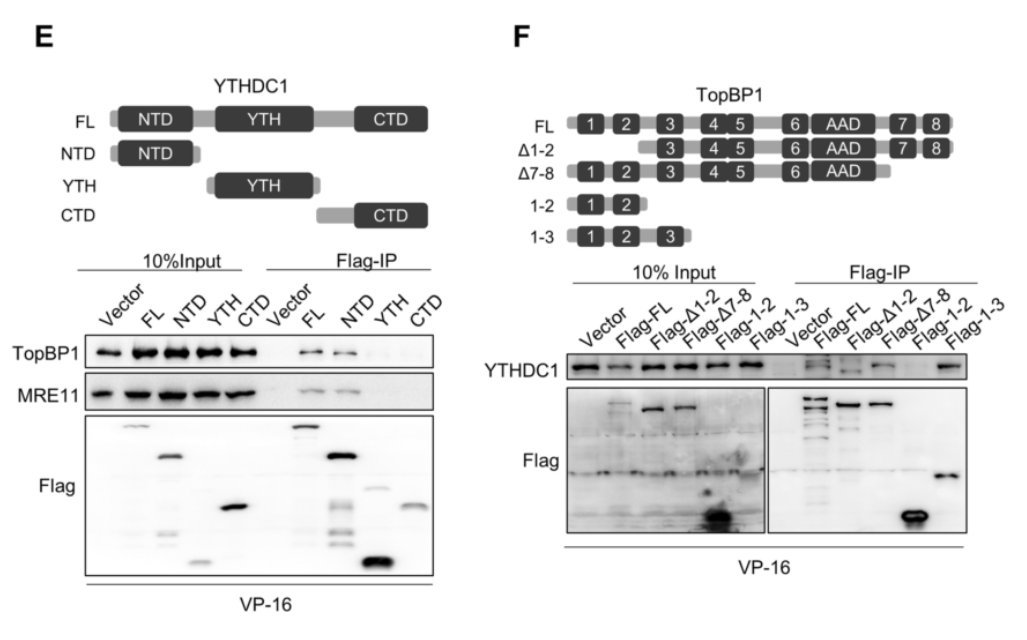
6. YTHDC1的缺失会导致基因组的不稳定
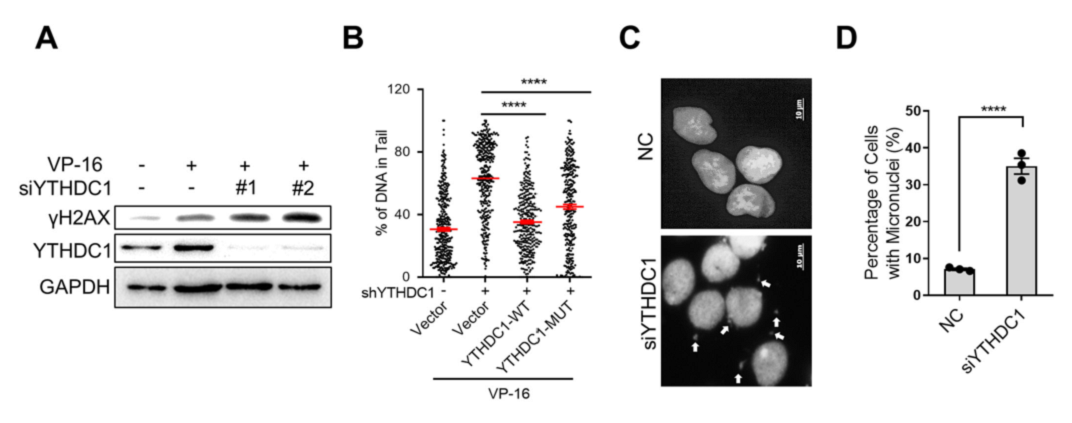
此前有研究报道,ATR激活失败会削弱DSB修复,导致DNA损伤积累和基因不稳定。图6A~B显示,在YTHDC1敲除的A549细胞中,γH2AX增加;彗星实验的结果显示,在YTHDC1敲除的细胞中有更多的DNA片段,而过表达YTHDC1野生组或YTHDC1突变组可以逆转这个情况。图6C~D显示,YTHDC1的缺失也增加了微核的水平,表明缺乏DNA损伤修复和基因组不稳定性的诱导。这些说明YTHDC1可以参与DNA损伤修复,以维持基因组学的稳定性。
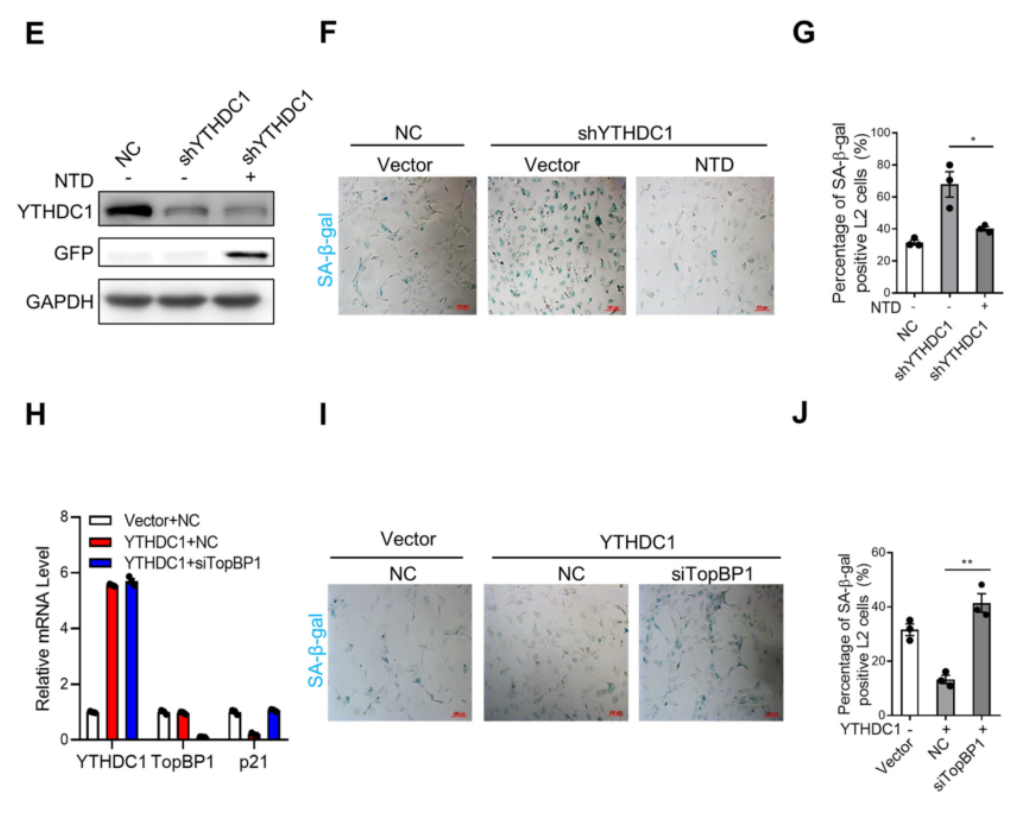
为此,作者还检测了YTHDC1是否通过TopBP1-ATR通路抵抗应激诱导的细胞衰老。图6E~G结果显示,在YTHDC1敲除的细胞中过表达YTHDC1的NTD结构域,发现该NTD结构域能够挽救ATR的激活,从而成功挽救了YTHDC1缺陷细胞的衰老。接着,我们从图6H~J可以看到,通过SA-β-gal染色和p21表达水平显示,TopBP1的缺失抵消了YTHDC1对衰老的保护作用。最终作者得出结论:YTHDC1通过NTD结构域与TopBP1结合,调节ATR的激活,在应激诱导的衰老中发挥保护作用。
四、研究小结
这篇文章揭示了YTHDC1通过促进TopBP1和MRE11之间的相互作用,从而激活ATR来及时修复DNA损伤和维持基因组的稳定性。此外,还揭示了YTHDC1在延缓细胞衰老方面的非典型作用,并指出提高肺中YTHDC1的表达可能是肺纤维化的有效治疗策略。
五、相关国自然中标项目分享

 文章推荐
文章推荐

业务咨询
业务咨询专线:133 7682 0615
Email:lxyjy@wie-biotech.com


